Ficha SCA5
Ataxia:
SCA5 (Ataxia Espinocerebelar Tipo 5)
GENES RELACIONADOS:
SPTBN2
LOCALIZAÇÃO:
Cromossoma 11 (11q13.2)
TIPO DE MUTAÇÃO:
SPTBN2 -> mutações pontuais, deleções e outras
ÚLTIMA ATUALIZAÇÃO:
01/04/2025 por Márcio Galvão
HERANÇA:
Autossômica Dominante
Conteúdo gerado com apoio de IA Generativa, revisado pelo autor.
1. SOBRE A SCA5
A Ataxia Espinocerebelar Tipo 5 (SCA5) é um tipo específico de ataxia dentro de um grupo de doenças hereditárias que afetam o sistema nervoso central. Na SCA5, defeitos genéticos causam problemas em fibras nervosas específicas que transportam mensagens para o cérebro e dele, resultando na degeneração do cerebelo (o centro de coordenação motora do cérebro) [1].
A SCA5 é uma doença progressiva causada por mutações no gene SPTBN2 (cromossomo 11q13.2), que codifica a proteína beta-III spectrina. A mutação foi mapeada para o cromossomo 11 em 1994 (Ranum et al), e em 2006 foram identificadas as mutações (deleções in-frame e mutações missense) que comprometem a estabilidade da proteína (Ikeda et al.). A proteína beta-III spectrina, juntamente com outras proteínas (alfa-spectrin, F-actin) formam o citoesqueleo que confere o formato e a resiliência mecânica para a membrana plasmática das células, o que é necessário para estabilizar e organizar outras proteínas que atuam em importantes processos celulares. Também estabiliza transportadores de glutamato e canais de cálcio nas membranas das células de Purkinje, neurônios cerebelares essenciais para a função motora (Perkins et al., 2010). A mutação no gene SPTBN2 que expressa esta proteína afeta o funcionamento normal e pode causar a morte destes neurônios (desregulação da sinalização intracelular e estresse do retículo endoplasmático), o que por consequência causa os sintomas da ataxia SCA5. Os interessados em informações mais técnicas podem consultar [4], [5], [8] e [9].
A SCA5 também é conhecida como ataxia de Holmes, devido ao fato de o Dr. Gordon Holmes ter sido o primeiro a descrever a doença em 1907, ou ainda ataxia de Lincoln, pois a SCA5 foi diagnosticada em descendentes (décima primeira geração) dos avós paternos presidente norte-americano Abraham Lincoln, embora não haja evidências diretas de que o próprio Lincoln fosse afetado [8]. Comparada com outras ataxias espinocerebelares como a SCA1, SCA2 e SCA3 a ataxia SCA5 progride mais lentamente e em geral não afeta a expectativa de vida do paciente [8].
Figura 1 - Diagrama gerado pelo autor com apoio de Inteligência Artificial
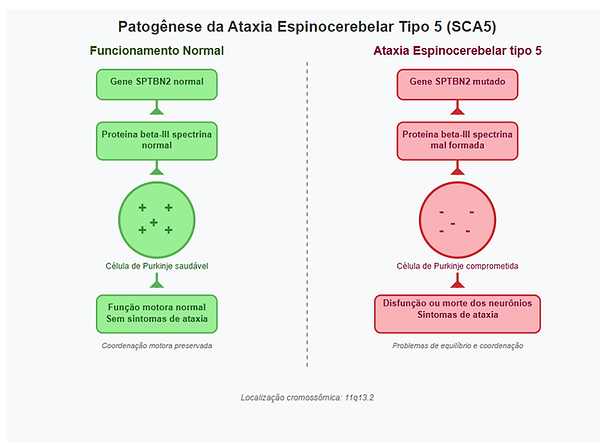
2. SINTOMAS TÍPICOS
A Ataxia Espinocerebelar Tipo 5 (SCA5) é classicamente definida como uma ataxia cerebelar "pura", pois suas manifestações clínicas estão predominantemente restritas à degeneração do cerebelo, estrutura responsável pela coordenação motora e equilíbrio. Os sintomas típicos da SCA5 são [5]:
-
A ataxia de marcha (desequilíbrio ao caminhar, dificuldade em subir escadas, e perda de equilíbrio ao ficar em um só pé por exemplo, durante o banho).
-
A falta de coordenação motora nos braços e mãos também é comum na SCA5, mas em geral não é tão desabilitante quanto na marcha. Os indivíduos afetados podem perceber uma deterioração na escrita manual, dificuldades ao abotoar camisas e outras atividades que requerem destreza manual.
-
Anormalidades oculomotoras (como o nistagmo).
Outras manifestações frequentes:
-
Disartria cerebelar: fala arrastada, porém raramente grave o suficiente para comprometer a comunicação oral.
-
Tremor de intenção (não em repouso).
Diferentemente de outras SCAs (como SCA3), a SCA5 raramente apresenta envolvimento significativo de regiões extra-cerebelares, como tronco encefálico ou vias autonômicas, preservando funções cognitivas, a deglutição, a digestão, o controle urinário e a força muscular. Desta forma, as pessoas com SCA5 normalmente continuam vivendo de forma independente [8]. Por exemplo, apesar de muitos pacientes com SCA5 eventualmente usarem algum auxílio para caminhar (como bengalas, ou andador), apenas raramente a doença chega ao ponto de trazer dependência de cadeira de rodas, como pode ocorrer em outras ataxias. Para mais informações sobre sintomas da SCA5 consultar [4, 5]
Os tipos de sintomas experimentados na ataxia SCA5 e sua severidade podem variar de pessoa para pessoa, mesmo dentro de uma mesma família.
3. IDADE DOS SINTOMAS
O surgimento dos sintomas (onset) da SCA5 geralmente ocorre na terceira ou quarta década de vida, mas há registros de ocorrência dos 10 aos 68 anos de idade [8]. A fonte [6] indica a idade média de surgimento dos sintomas como 33 anos, com variação entre 6 e 68 anos.
Expansão do espectro clínico
Inicialmente descrita como uma doença de início exclusivamente adulto, estudos genéticos posteriores identificaram mutações específicas no gene SPTBN2 associadas a manifestações precoces (infância ou adolescência), que estão ligadas a fenótipos mais graves, incluindo:
-
Atraso no desenvolvimento motor e cognitivo, sugerindo envolvimento extra-cerebelar (ex.: córtex cerebral ou vias subcorticais) (Avery et al., 2020). Vale ressaltar que o envolvimento cognitivo em casos pediátricos é uma exceção, e não a regra. A SCA5 é predominantemente uma ataxia cerebelar "pura".
-
Progressão acelerada de sintomas, como ataxia de marcha incapacitante e disartria marcante (Hoxha et al., 2022).
Esse fenótipo expandido desafia a noção clássica da SCA5 como uma ataxia cerebelar "pura", indicando que mutações severas no SPTBN2 podem afetar redes neurais além do cerebelo, ainda que de forma limitada (Paulson et al., 2023).
Até 2023, mais de 20 variantes patogênicas do gene SPTBN2 que podem causar a SCA5 já foram reportadas, abrangendo mutações pontuais, deleções e variantes de splicing [8, 9].
4. ANTECIPAÇÃO
Análises de pacientes (relatos observacionais) mostraram que filhas tiveram sintomas de 10 a 20 anos mais cedo do que suas mães, que também tinham a doença, e estas por sua vez apresentaram sintomas mais cedo que as suas mães, as avós das netas [5]. Apesar disso, não há evidências moleculares que sustentem a ocorrência de antecipação verdadeira na SCA5 (Paulson et al., 2023). Ou seja, a "antecipação" na SCA5 nos casos relatados é aparente, e não genética.
A antecipação clínica—fenômeno em que os sintomas surgem progressivamente mais cedo em gerações subsequentes—é uma característica clássica de doenças por expansão de repetições de trinucleotídeos (ex.: SCA1, SCA2, SCA3). Entretanto, a SCA5 envolve mutações não repetitivas no gene SPTBN2 (deleções in-frame ou mutações missense), e neste caso os mecanismos genéticos convencionais da antecipação (ex.: expansão de repetições) estariam ausentes. Neste contexto, é possível que os relatos de famílias com aparente antecipação (ex.: sintomas 10-20 anos mais precoces em netas vs. avós) sejam atribuídos a outros fatores (facilidade de diagnóstico, modificadores epigenéticos ou ambientais, ou variantes diferentes do SPTBN2 coexistindo na mesma linhagem familiar.
Nos casos em que o surgimento dos sintomas ocorre na idade adulta a progressão da ataxia é mais lenta [8].
5. HERANÇA
A SCA5 é uma doença com herança autossômica dominante. Isto significa que indivíduos de ambos os sexos têm a mesma probabilidade de herdar uma cópia (alelo) do gene com mutação e se tornarem portadores da mutação. Um filho de uma pessoa com SCA5 tem uma probabilidade de 50% de herdar uma cópia do gene alterado (considerando que apenas um dos pais é portador da mutação, ou seja, a mãe ou o pai biológico).
Nota: "Autossômica" significa que o gene está localizado em qualquer cromossoma com exceção dos cromossomas sexuais X e Y. Os genes, assim como os cromossomas, normalmente existem em pares (temos um par de cada gene, uma cópia do gene é herdada da mãe, outra do pai). "Dominante" significa que apenas uma cópia do gene responsável (um alelo) herdada do pai ou da mãe é suficiente para transmitir uma característica física (como a presença de covinhas nas bochechas), ou uma doença genética (como as ataxia hereditárias) de uma geração (pais) para a seguinte (filhos).
Figura 2 - Fonte: MedlinePlus, U.S. National Library of Medicine.

Nota: Embora a SCA5 tenha transmissão autossômica dominante, mutações homozigóticas do gene SPTBN2 podem causar um outro tipo de ataxia espinocerebelar com transmissão autossômica recessiva, codificada como SCAR14, que usualmente tem sintomas mais severos e com surgimento (onset) mais cedo, atraso no desenvolvimento e problemas cognitivos. Uma mutação homozigótica ocorre quando a pessoa herda duas cópias do mesmo gene com mutação, uma do pai biológico e outra da mãe. Em contraste, a mutação heterozigótica ocorre quando a pessoa herda apenas um alelo (cópia do gene) com mutação, sendo o outro alelo normal. Consultar [5, 8] para mais informações.
6. PREVALÊNCIA
A SCA5 é uma ataxia rara, mesmo entre as ataxias com transmissão dominante. A prevalência é desconhecida, mas estima-se que seja de menos que 1 caso em cada 100.000 de habitantes [6]. Famílias nos Estados Unidos, França, Alemanha, Japão, Noruega e outros países já foram reportadas com com SCA5.
7. INFORMAÇÕES ADICIONAIS
Diagnóstico - O diagnóstico da SCA5 pode ser feito com teste genético molecular (exame de DNA) para detectar rearranjos e mutações pontuais em heterozigose no gene SPTBN2, sendo recomendado sobretudo se já houver alguém na família com diagnóstico fechado (histórico familiar positivo para SCA5). Antes da solicitação de testes genéticos, o neurologista tipicamente faz exames clínicos neurológicos para análise de sintomas, reflexos, anormalidades oculares, avaliação de histórico familiar etc. e é normal que solicite exames de imagem para verificar se há atrofia cerebelar por exemplo (Figura 3),
Nota: Embora o diagnóstico por teste genético possa ser difícil, demorado e caro, ele é importante, pois permite um melhor aconselhamento genético para os familiares (risco de transmissão da mutação para futuras gerações na família), um melhor manejo da doença, que estará bem determinada, e também possibilita a participação do paciente em ensaios clínicos para medicamentos para ataxias específicas.
Figura 3 - NAF (National Ataxia Foundation) webinar "Research and Treatment Development for SCA5" with Dr. Adam Avery [9].

8. TERAPIAS E MEDICAMENTOS EM TESTES PARA ESTA ATAXIA
Ver artigo "Reimagining drugs for rare brain disorder" por Marissa Locke Rottinghaus, Feb. 16, 2023
A fonte [9] lista algumas abordagens terapêuticas possíveis para a SCA5 (ainda em estudos):
-
Edição genética (de variates pagogênicas do gene SPTBN2) por CRISPR/Cas9.
-
Terapias ASO (Antisense Oligonucleotides) para reduzir expressividade da proteína mutante beta-III spectrin.
-
Novos medicamentos para tratar os efeitos (ex. problemas na sinalização do neurotransmissor glutamato)
9. TRATAMENTOS
A ataxia SCA5 ainda não tem cura, mas é possível tratar sintomas visando melhor qualidade de vida e fornecer apoio contínuo ao paciente. É importante que os pacientes com SCA5 sejam acompanhados por um neurologista e uma equipe médica multidisciplinar e especializada, com a inclusão gradual de novos profissionais de saúde na medida em que seja necessário em função dos sintomas (geneticista, neuro oftalmologista, fisioterapeuta neurofuncional, fisioterapeuta ocupacional, fonoaudiólogo, nutricionista etc.).
Seguem algumas recomendações gerais para o manejo de sintomas na SCA5:
-
Fisioterapia neurofuncional, exercícios (sobretudo bicicleta ergométrica) e outras atividades físicas regulares (Yoga, Pilates, hidroginástica etc.) são recomendados (dentro das possibilidades de cada um).
-
Para reduzir o risco de quedas em função das dificuldades de equilíbrio ao caminhar pode-se adotar bengalas, andadores ou (bem mais raramente), cadeira de rodas, dependendo do estágio da doença.
-
Fisioterapia ocupacional e algumas adaptações em casa e nos hábitos diários podem ajudar (ex. instalar barras de apoio nos corredores e banheiros, cadeira para banho, luzes para iluminação noturna, reposicionar os móveis para facilitar a mobilidade, remover tapetes para não tropeçar, utilizar copos com tampa e canudo, calçados com sola antiderrapante e fáceis de calçar etc.).
-
Descansar sempre que necessário, e é importante ter um sono noturno de boa qualidade. No caso de dificuldades para dormir, consultar o médico, pois há medicamentos que podem ajudar (por exemplo, óleo de Canabidiol).
-
Manter uma alimentação saudável e com boa hidratação.
-
Suplementos e vitaminas podem ser recomendados (consultar o médico para avaliar a necessidade - não consumir vitaminas e suplementos sem supervisão médica).
-
Convém controlar o peso para evitar dificuldades ainda maiores na mobilidade.
-
Para a disartria, se tal sintoma se manifestar, recomenda-se terapia especializada de fala (fonoaudiologia). Dependendo do estágio pode-se avaliar o uso de dispositivos de assistência para a comunicação (disponíveis para smartphone, computador, iPad etc.).
-
Evitar (tanto quanto possível) o estresse, que em geral piora os sintomas da ataxia.
-
Se necessário, há medicações para o manejo da ansiedade e da depressão. Procurar o médico para avaliar as alternativas mais adequadas.
Nota! Alguns pacientes com ataxias cerebelares diversas relatam benefícios e melhoria de sintomas de ataxia após sessões de neuromodulação ou estimulação cerebelar não invasiva, por exemplo, a estimulação transcraniana por corrente contínua (tDCS), ou estimulação magnética transcraniana (TMS) com fisioterapeutas certificados. Observar que embora já esteja sendo comercializada esta terapia ainda não foi aprovada pela FDA nos Estados Unidos (ou pela ANVISA no Brasil) para tratamento de ataxias (ou seja, trata-se de tratamento experimental e sem garantias).
Veja informações sobre medicamentos para sintomas de ataxias.
Veja informações sobre tratamentos e cuidados para os pacientes.
Veja informações para quem tem diagnóstico recente.
Veja informações sobre Grupos de Suporte para pacientes e cuidadores.
10. REFERÊNCIAS
As referências abaixo englobam fontes acadêmicas e de organizações especializadas que embasaram as informações desta ficha técnica, incluindo artigos revisados por pares, repositórios genéticos – OMIM, resumos de literatura – GeneReviews, e materiais informativos de fundações de ataxia. Para mais informações, consulte a lista de Referências do ataxia.info.
Ref #1
Fonte:
NAF (National Ataxia Foundation)
© Copyright National Ataxia Foundation
Idioma:
Inglês
Data:
Revised NAF– 01/2019
Ref #2
Fonte:
GARD - Genetic and Rare Diseases Information Center.
Copyright © National Center for Advancing Translational Sciences - National Institutes of Health (NIH) ©.
Idioma:
Inglês
Data:
Last Updated: November 2023
Ref #3
Fonte:
NEUROMUSCULAR DISEASE CENTER (Alan Pestronk, MD)
Washington University, St. Louis, MO - USA
Idioma:
Inglês
Data:
Last Updated: Please see https://neuromuscular.wustl.edu/rev.htm
Ref #4
Fonte:
Katherine A. Dick, Yoshio Ikeda, John W. Day, Laura P.W. Ranum
ScienceDirect Copyright © 2024 Elsevier B.V.
Idioma:
Inglês
Data:
2012
Ref #5
Fonte:
OMIM® - An Online Catalog of Human Genes and Genetic Disorders.
Copyright © Johns Hopkins University.
Idioma:
Inglês
Data:
Edit History: carol : 04/02/2021
Ref #6
Fonte:
Expert reviewer(s): Dr Shinsuke FUJIOKA - Dr Yoshio TSUBOI - Dr Zbigniew WSZOLEK
Copyright ® Orphanet - The portal for rare diseases and orphan drugs
Idioma:
Inglês.
Data:
Last update: February 2020
Ref #7
Fonte:
Sarah Denha
YouTube - Copyright ® Oakland University
Idioma:
Inglês.
Data:
Nov 12, 2021
Ref #8
Fonte:
Presented by: Dr. Theresa Zesiewicz
YouTube - Copyright © National Ataxia Foundation (NAF)
Idioma:
Inglês. É possível habilitar legendas e configurar tradução automática das legendas para português.
Data:
March 8th, 2024
Ref #9
Fonte:
Presented by: Dr. Adam Avery
YouTube - Copyright © National Ataxia Foundation (NAF)
Idioma:
Inglês. É possível habilitar legendas e configurar tradução automática das legendas para português.
Data:
March 19th, 2024